


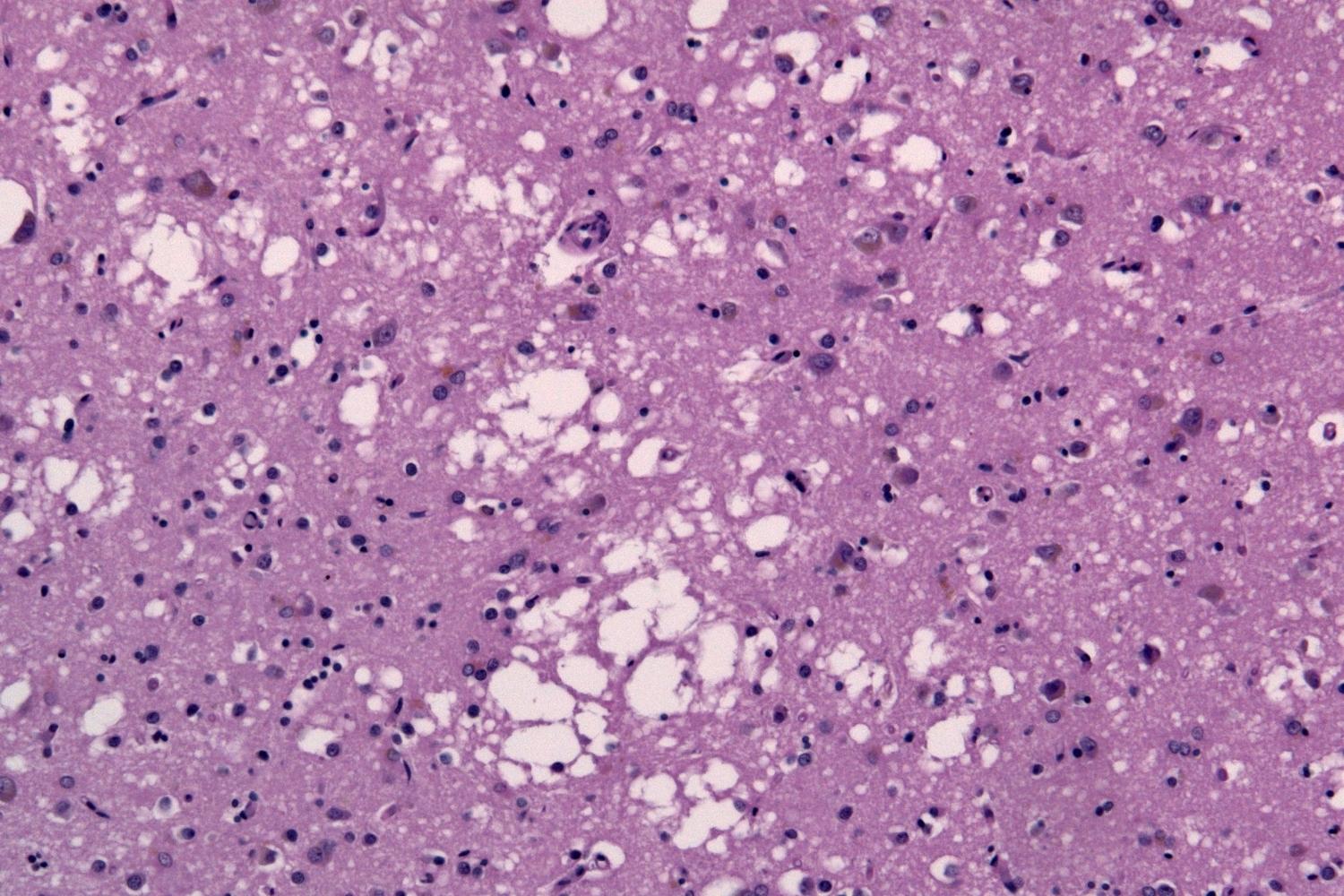
Jakob–Creutzfeldt Disease (JCD) is a rare, fatal brain disorder that affects about one in a million people each year. This disease falls under the category of transmissible spongiform encephalopathies (TSEs), which cause the brain to develop sponge-like holes. Symptoms often start with memory problems, personality changes, and difficulties with movement. As the disease progresses, it leads to severe mental impairment and physical disabilities. JCD can be sporadic, inherited, or acquired through exposure to infected brain tissue. There is no cure, and treatment focuses on alleviating symptoms. Understanding the facts about Jakob–Creutzfeldt Disease can help raise awareness and support ongoing research efforts.
Key Takeaways:
- CJD is a rare brain disease caused by abnormal proteins. It has no cure and can be challenging to diagnose. Research is ongoing to find treatments and prevent its spread.
- CJD has different types and can be hereditary, acquired, or sporadic. It's not contagious through casual contact, but medical procedures and contaminated beef products can transmit the disease.
What is Jakob–Creutzfeldt Disease?
Jakob–Creutzfeldt Disease (CJD) is a rare, degenerative brain disorder. It affects about one in a million people each year worldwide. Here are some intriguing facts about this mysterious disease.
- CJD is caused by abnormal proteins called prions, which can trigger normal proteins in the brain to fold abnormally.
- There are three main types of CJD: sporadic, hereditary, and acquired.
- Sporadic CJD accounts for about 85% of cases and occurs with no known cause.
- Hereditary CJD is linked to a genetic mutation and makes up about 10-15% of cases.
- Acquired CJD can result from exposure to infected brain or nervous system tissue, often through medical procedures.
Symptoms and Diagnosis
Recognizing CJD can be challenging due to its varied symptoms. Here are some key points about its symptoms and diagnosis.
- Early symptoms often include memory problems, behavioral changes, and coordination issues.
- As the disease progresses, patients may experience severe mental deterioration, muscle stiffness, and involuntary movements.
- Diagnosis typically involves a combination of neurological exams, MRI scans, and cerebrospinal fluid tests.
- A definitive diagnosis can only be made through a brain biopsy or autopsy.
- EEG tests can sometimes show characteristic patterns associated with CJD.
Transmission and Risk Factors
Understanding how CJD spreads and who is at risk can help in managing and preventing the disease.
- CJD is not contagious through casual contact.
- Prions can survive standard sterilization procedures, making medical transmission possible.
- Consuming contaminated beef products has been linked to a variant form of CJD known as vCJD.
- Family history of prion diseases increases the risk of hereditary CJD.
- Certain medical procedures, such as corneal transplants or use of contaminated surgical instruments, can transmit CJD.
Treatment and Prognosis
Currently, there is no cure for CJD, but understanding treatment options and prognosis is crucial.
- Treatment focuses on alleviating symptoms and providing supportive care.
- Medications like anticonvulsants can help manage muscle spasms and seizures.
- Most patients succumb to the disease within a year of symptom onset.
- Research is ongoing to find potential treatments and a cure for prion diseases.
- Early diagnosis can help in managing symptoms more effectively.
Historical and Notable Cases
CJD has a fascinating history and has affected some notable individuals.
- The disease was first described by German neurologists Hans Gerhard Creutzfeldt and Alfons Maria Jakob in the 1920s.
- Nobel Prize-winning scientist Stanley B. Prusiner discovered prions, the infectious agents causing CJD, in 1982.
- British cattle herds experienced a significant outbreak of Bovine Spongiform Encephalopathy (BSE), or "mad cow disease," in the 1980s, leading to cases of vCJD in humans.
- The first known case of vCJD was identified in the United Kingdom in 1996.
- Notable individuals, including professional athletes and musicians, have been diagnosed with CJD, bringing more attention to the disease.
Research and Future Directions
Ongoing research aims to better understand and combat CJD. Here are some current and future directions in the field.
- Scientists are exploring ways to detect prions in blood and other tissues before symptoms appear.
- Gene therapy is being investigated as a potential treatment for hereditary CJD.
- Animal models, such as mice and sheep, are used to study prion diseases and test new treatments.
- Public health initiatives focus on preventing the spread of prion diseases through improved sterilization techniques and food safety measures.
- International collaboration among researchers and healthcare professionals is crucial for advancing our understanding of CJD and finding a cure.
Final Thoughts on Jakob–Creutzfeldt Disease
Jakob–Creutzfeldt Disease (JCD) is a rare, fatal brain disorder that affects about one in a million people each year. Caused by abnormal proteins called prions, JCD leads to rapid mental deterioration, memory loss, and motor dysfunction. There are no known cures, and treatments focus on alleviating symptoms and providing supportive care. Early diagnosis is challenging due to its similarity to other neurological conditions. Research continues to explore potential therapies and better diagnostic tools. Awareness and understanding of JCD are crucial for early intervention and support for affected individuals and their families. While JCD remains a medical mystery in many ways, ongoing studies offer hope for future breakthroughs. Stay informed and support research efforts to combat this devastating disease.
Frequently Asked Questions
Was this page helpful?
Our commitment to delivering trustworthy and engaging content is at the heart of what we do. Each fact on our site is contributed by real users like you, bringing a wealth of diverse insights and information. To ensure the highest standards of accuracy and reliability, our dedicated editors meticulously review each submission. This process guarantees that the facts we share are not only fascinating but also credible. Trust in our commitment to quality and authenticity as you explore and learn with us.