


What is Osler-Weber-Rendu Disease? Osler-Weber-Rendu Disease, also known as Hereditary Hemorrhagic Telangiectasia (HHT), is a rare genetic disorder that affects blood vessels. This condition leads to abnormal blood vessel formation, causing frequent nosebleeds, red spots on the skin, and more severe complications like arteriovenous malformations (AVMs) in organs such as the lungs, liver, and brain. Inherited in an autosomal dominant pattern, each child of an affected parent has a 50% chance of inheriting the disease. Early diagnosis and management are crucial for improving quality of life and preventing life-threatening complications. Understanding the symptoms, genetic basis, and treatment options can help those affected lead near-normal lives.
Key Takeaways:
- Osler-Weber-Rendu Disease, also known as HHT, is a rare genetic disorder causing abnormal blood vessel formation. It can lead to nosebleeds, skin spots, and serious complications in organs like the lungs and brain.
- Early diagnosis and management are crucial for HHT. Regular screening and a multidisciplinary care team can help prevent complications and improve quality of life for those affected by this complex genetic disorder.
What is Osler-Weber-Rendu Disease?
Osler-Weber-Rendu disease, also known as hereditary hemorrhagic telangiectasia (HHT), is a rare genetic disorder affecting blood vessels. This condition can lead to various complications due to abnormal blood vessel formation. Here are some key facts to help you understand this complex disease.
-
Definition and Synonyms: Osler-Weber-Rendu disease is also called hereditary hemorrhagic telangiectasia (HHT). It involves abnormal blood vessel formation, leading to bleeding and blood flow issues.
-
History: First described by Henry Gawen Sutton in 1864, it was later distinguished from hemophilia by Henri Jules Louis Marie Rendu in 1896. William Osler and Frederick Parkes Weber further characterized it in the early 1900s.
-
Inheritance Pattern: HHT is inherited in an autosomal dominant manner. This means a single copy of the mutated gene from an affected parent can cause the disease.
-
Prevalence: HHT affects over a million people globally. It is often underdiagnosed due to its varied symptoms and lack of specific diagnostic criteria until recent years.
Genetic Basis and Clinical Manifestations
Understanding the genetic basis and clinical signs of HHT is crucial for early diagnosis and management. Here are some important points.
-
Genetic Basis: The most common genes involved are endoglin (ENG) and activin receptor-like kinase type I (ALK-1). Mutations in these genes disrupt normal blood vessel development.
-
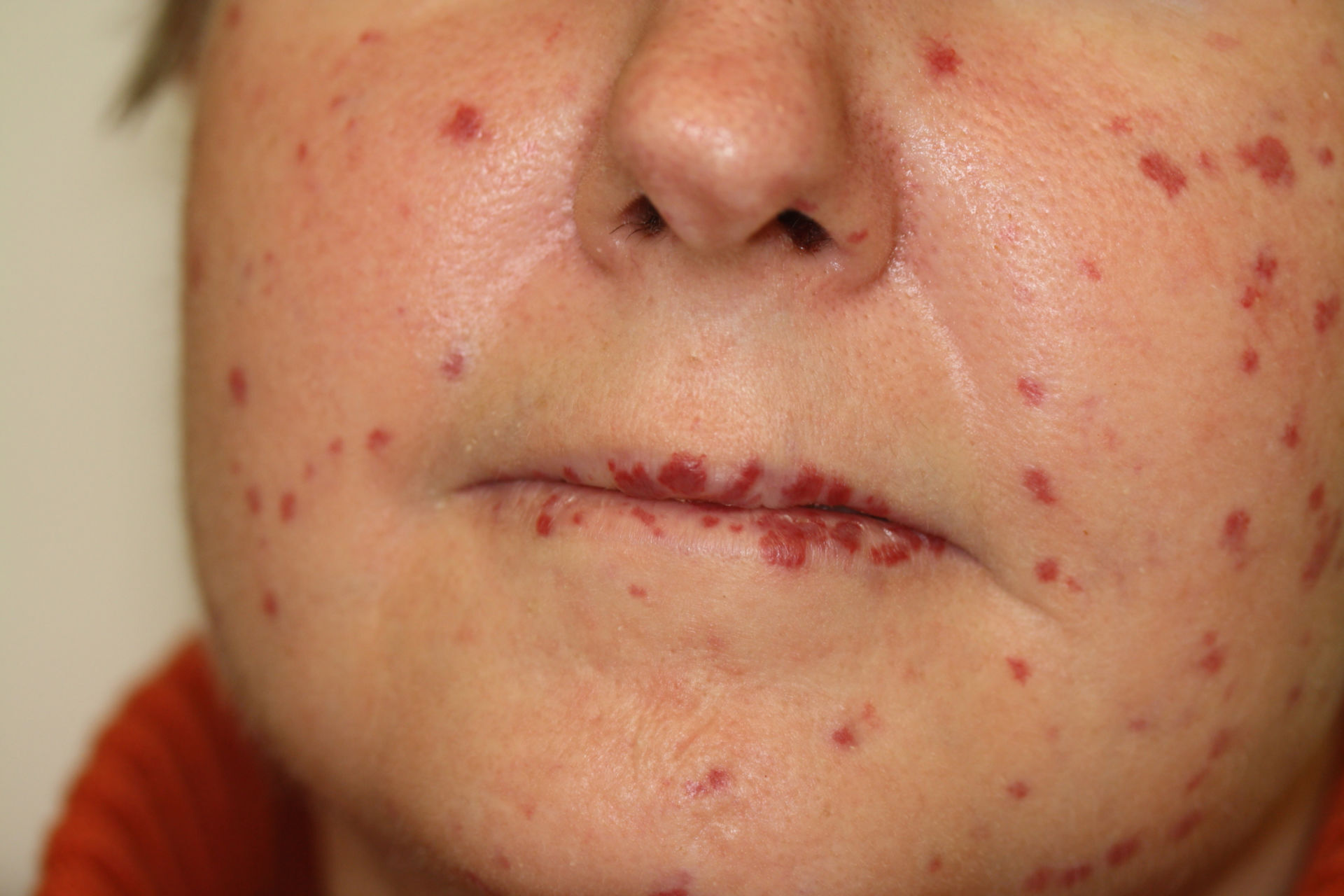
Clinical Manifestations: The earliest sign is often recurrent nosebleeds (epistaxis). Telangiectasias, or small dilated blood vessels, usually appear on the skin and mucous membranes by the third decade of life.
-
Telangiectasia: These small, dilated blood vessels appear as red spots or patches on the skin and mucous membranes, particularly on the nose, lips, tongue, and gastrointestinal tract.
-
Epistaxis: Frequent nosebleeds are the most common symptom. Severe cases can lead to significant anemia, requiring transfusions.
-
Gastrointestinal Bleeding: This is another common issue, leading to anemia and sometimes necessitating iron or blood transfusions.
Visceral Involvement and Complications
HHT can affect various organs, leading to serious complications. Here are some facts about visceral involvement and potential complications.
-
Visceral Involvement: HHT can affect organs like the lungs, liver, and brain. Arteriovenous malformations (AVMs) in these organs can cause severe complications.
-
Pulmonary AVMs: These are common in HHT and can lead to significant health issues. They increase the risk of stroke and brain abscess by allowing thrombotic and septic emboli to bypass the pulmonary vasculature.
-
Central Nervous System (CNS) Involvement: CNS involvement can manifest as migraines, seizures, and focal neurologic symptoms. Stroke and brain abscess are more common due to the loss of normal pulmonary vasculature filtering function.
-
Spinal AVMs: Rarely, HHT can cause spinal AVMs, mainly affecting children. This can lead to acute paraplegia due to spinal arteriovenous fistula.
Diagnosis and Screening
Early diagnosis and screening are vital for managing HHT effectively. Here are some key points about the diagnostic process.
-
Diagnostic Criteria: The Curaçao criteria, established in 1999, are used for diagnosing HHT. These include epistaxis, telangiectasias, visceral lesions, and family history.
-
Classification: Diagnosis is classified as definite if three or four criteria are present, possible or suspected if two criteria are present, and unlikely if fewer than two criteria are present.
-
Family Screening: Screening family members includes a complete history, physical examination, chest radiography, and arterial blood gas testing.
-
Genetic Testing: This can confirm mutations in genes like ENG and ALK-1. It is useful for identifying carriers who may not yet show symptoms.
Management and Treatment
Managing HHT involves both medical and surgical options. Here are some important aspects of treatment.
-
Management of Bleeding: Treatments include tranexamic acid for nosebleeds and bevacizumab for severe bleeding episodes. Surgical interventions may be necessary for AVMs.
-
Surgical Management: This involves embolization or resection of AVMs, significantly reducing the risk of bleeding and other complications.
-
Prognosis: Prognosis varies depending on symptom severity. With proper screening and management, life expectancy can be near-normal. Untreated patients face higher risks of complications like stroke and brain abscess.
-
Quality of Life: Recurrent bleeding and complications can impact quality of life. However, appropriate treatment allows many patients to lead near-normal lives.
Complications and Associated Conditions
HHT can lead to various complications and associated conditions. Here are some key points to be aware of.
-
Complications: These include anemia, gastrointestinal bleeding, pulmonary hypertension, liver dysfunction, and intracranial hemorrhages. Early diagnosis and management are crucial.
-
Migraine Headaches: Migraines occur in 13-50% of HHT patients. The reason for this association is unclear but is more common in those with pulmonary AVMs.
-
Neurologic Involvement: This includes migraines, seizures, and focal neurologic symptoms. Stroke and brain abscess are more common due to the loss of normal pulmonary vasculature filtering function.
Screening Recommendations and Chronic Management
Regular screening and chronic management are essential for preventing complications and improving quality of life. Here are some recommendations.
-
Screening Recommendations: Family members should undergo a complete history, physical examination, chest radiography, and arterial blood gas testing for early detection.
-
Treatment Indications: Treatment varies based on the site of involvement and presentation. Mild cases may not require treatment, while severe cases need medical and surgical management.
-
Coordination of Care: A multidisciplinary team approach is recommended, involving hematologists, radiologists, surgeons, and other specialists.
-
Chronic Management: Regular follow-up appointments help monitor for signs of bleeding or other complications, preventing life-threatening episodes and improving quality of life.
Research and Awareness
Ongoing research and raising awareness are crucial for improving diagnosis and treatment. Here are some final points.
-
Research and Guidelines: Recent studies have led to new guidelines for diagnosing and managing HHT. The Second International Guidelines, published in 2020, emphasize early detection and aggressive management.
-
Awareness and Education: Raising awareness about HHT is vital for early diagnosis and management. Educational programs and support groups help families understand the condition and seek appropriate care.
Final Thoughts on Osler-Weber-Rendu Disease
Osler-Weber-Rendu disease, or hereditary hemorrhagic telangiectasia (HHT), is a rare but impactful condition. It affects blood vessels, leading to telangiectasias and arteriovenous malformations (AVMs). These can cause frequent nosebleeds, gastrointestinal bleeding, and serious complications in organs like the lungs, liver, and brain. Early diagnosis using the Curaçao criteria and genetic testing is crucial. Management involves both medical and surgical treatments to control bleeding and prevent complications. Regular screening and a multidisciplinary approach can significantly improve the quality of life for those affected. Awareness and education about HHT are essential for early detection and effective management. With proper care, individuals with HHT can lead near-normal lives, minimizing the risk of life-threatening issues.
Frequently Asked Questions
Was this page helpful?
Our commitment to delivering trustworthy and engaging content is at the heart of what we do. Each fact on our site is contributed by real users like you, bringing a wealth of diverse insights and information. To ensure the highest standards of accuracy and reliability, our dedicated editors meticulously review each submission. This process guarantees that the facts we share are not only fascinating but also credible. Trust in our commitment to quality and authenticity as you explore and learn with us.