
What is Papillorenal Syndrome? Papillorenal Syndrome, also known as Renal Coloboma Syndrome, is a rare genetic disorder that affects the development of kidneys and eyes. This condition is caused by mutations in the PAX2 gene, which plays a crucial role in the early development of these organs. Individuals with this syndrome often experience underdeveloped kidneys (renal hypoplasia) and abnormalities in the optic nerve, leading to vision problems. The syndrome is inherited in an autosomal dominant manner, meaning a single copy of the mutated gene can cause the disorder. Symptoms can vary widely, making early diagnosis and management essential.
Key Takeaways:
- Papillorenal Syndrome (PRS) is a rare genetic disorder affecting kidneys and eyes, caused by mutations in the PAX2 gene. It can lead to severe complications like end-stage renal disease and visual impairment.
- Understanding the role of the PAX2 gene, risk factors, and potential complications of PRS is crucial for early diagnosis and effective management. Ongoing research aims to improve therapeutic interventions and management strategies.
What is Papillorenal Syndrome?
Papillorenal syndrome (PRS), also known as renal coloboma syndrome, is a rare genetic disorder that affects the development of kidneys and eyes. This condition can lead to severe complications, including end-stage renal disease and visual impairment. Let's dive into some key facts about this syndrome.
-
Definition and Synonyms: PRS is an autosomal dominant disorder impacting kidney and eye development. It's also known as renal coloboma syndrome, PAX2-related disorder, optic nerve coloboma with renal disease, coloboma-ureteral-renal syndrome, and optic coloboma, vesicoureteral reflux, and renal anomalies syndrome.
-
Prevalence: The exact prevalence of PRS is unknown, but at least 60 cases have been reported in scientific literature. Approximately 268 affected individuals are recorded in The Human Variome Project PAX2 database.
-
Genetic Cause: PRS is caused by variants (mutations) in the PAX2 gene, which provides instructions for making a protein involved in the early development of the eyes, ears, brain, and spinal cord. The PAX2 gene is located on chromosome 10q24.
-
Inheritance Pattern: PRS is inherited in an autosomal dominant fashion, meaning a single copy of the mutated gene is sufficient to cause the condition. However, about 65% of patients with PRS and PAX2 mutations do not have a family history, suggesting de novo mutations or parental germline mosaicism.
Clinical Manifestations of Papillorenal Syndrome
PRS presents with a variety of symptoms primarily affecting the kidneys and eyes. Understanding these manifestations can help in early diagnosis and management.
-
Kidney Abnormalities: Renal hypoplasia (underdeveloped kidneys) can lead to end-stage renal disease (ESRD), where kidneys fail to filter fluids and waste products effectively. Other kidney issues include vesicoureteral reflux (backflow of urine from the bladder), multiple kidney cysts, and loose joints.
-
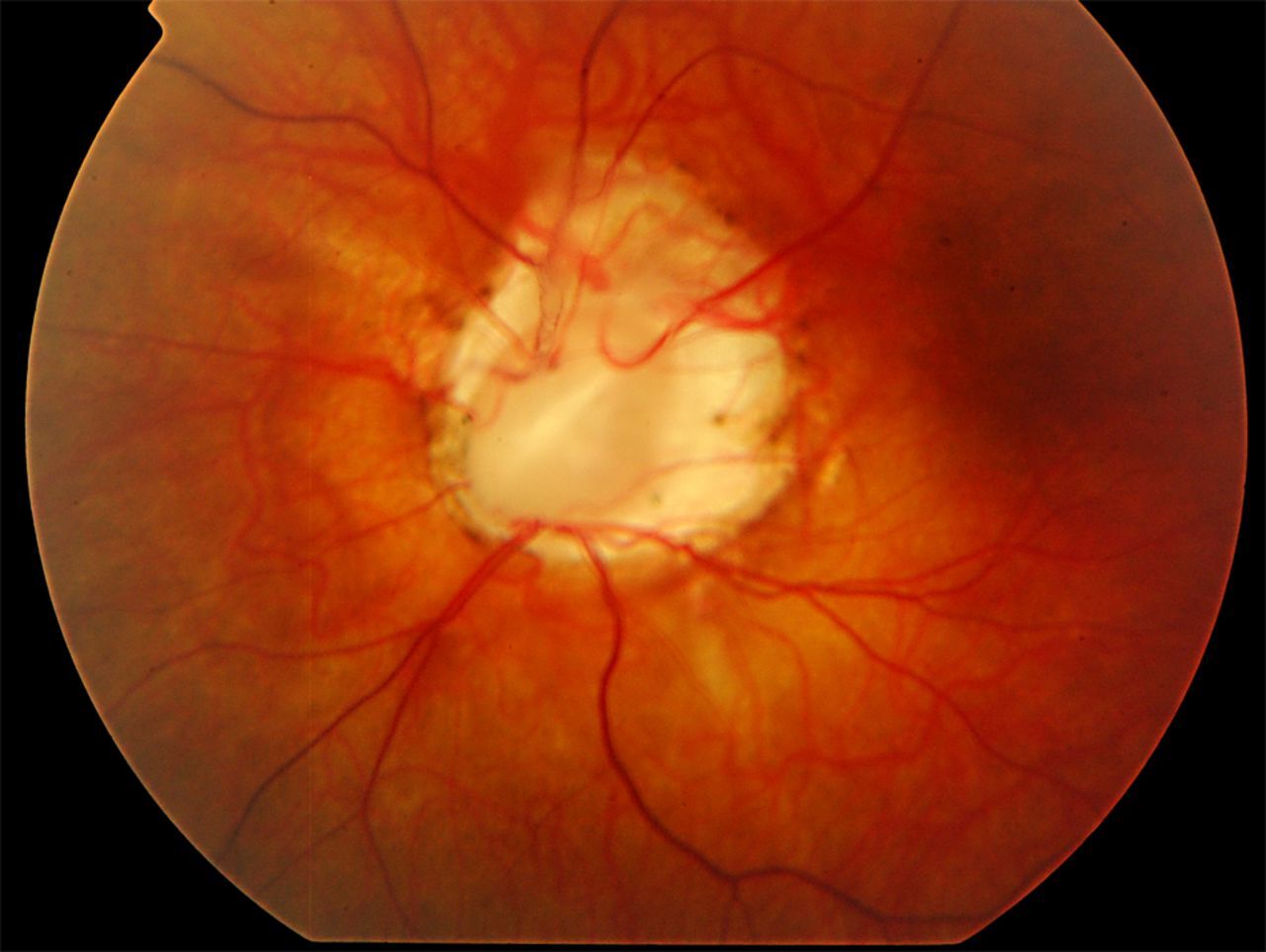
Eye Abnormalities: Optic nerve malformations can result in a gap or hole (coloboma) in the retina. Vision problems vary depending on the size and location of the malformation, ranging from no visual problems to severely impaired vision.
-
Additional Features: Less common features of PRS include high-frequency hearing loss and CNS abnormalities such as Chiari 1 malformations. Some patients may also experience retinal detachment and blindness due to eye malformations.
The Role of the PAX2 Gene
The PAX2 gene plays a crucial role in the development of various organs. Mutations in this gene are central to the development of PRS.
-
PAX2 Gene Function: The PAX2 protein is a transcription factor involved in altering gene expression for the early development of the eyes, kidneys, ears, brain, and spinal cord. Post-birth, the PAX2 protein continues to mediate cellular stress responses, apoptosis signaling processes, and vascular development.
-
Mutation Types: PAX2 gene mutations in PRS can cause either haploinsufficiency (complete loss of protein function in one allele) or missense mutations (partial or abnormal protein function). These mutations are inherited in a heterozygous autosomal dominant fashion, with homozygous genotypes being perinatal lethal.
-
De Novo Mutations: About 65% of patients with PRS and PAX2 mutations do not have a family history, suggesting that de novo mutations or parental germline mosaicism can also cause PRS.
Risk Factors and Complications
Understanding the risk factors and potential complications of PRS can help in managing the condition more effectively.
-
Teratogen Exposure: The presence of teratogens during pregnancy can increase the risk for multi-organ malformations, especially during the embryonic period (postfertilization weeks 3 to 8).
-
Systemic Diseases: Systemic diseases that compromise kidney or eye structures and functions (such as hypertension, hypercholesterolemia, and hyperglycemia) can increase the risk for vascular dysgenesis and worsen the prognosis.
-
Complications and Prognosis: The complications associated with PRS can lead to severe outcomes, including end-stage renal disease and visual impairment. The prognosis is generally poor, especially if systemic diseases compromise kidney or eye functions.
Diagnosis and Symptoms
Early diagnosis and understanding the symptoms of PRS are crucial for effective management and treatment.
-
Diagnosis: The diagnosis of PRS is primarily based on genetic testing for mutations in the PAX2 gene. Imaging studies such as ultrasound and MRI may also be used to confirm kidney and eye abnormalities.
-
Symptoms: Symptoms of PRS can vary widely among individuals but often include failure to thrive in infancy and early childhood, multicystic renal dystrophy, hypoplastic kidneys, vesicoureteral reflux, morning glory anomaly, coloboma of the optic disc, and progressive renal failure.
Genetic Analysis and Pregnancy Complications
Genetic analysis and understanding pregnancy complications can provide insights into the development and management of PRS.
-
Genetic Analysis: Genetic analysis has revealed that mutations in the PAX2 gene are associated with PRS. For example, a mutation of the PAX2 gene (619 insG) has been identified in a patient with PRS.
-
Pregnancy Complications: Exposure to teratogens during pregnancy, such as beta-interferon treatment for multiple sclerosis, can influence the manifestation or severity of the PAX2 mutant phenotype in children.
Risk Factors and Mutation Frequency
Identifying risk factors and understanding the frequency of PAX2 mutations can help in early detection and management of PRS.
-
Risk Factors: The greatest risk factor for developing PRS is a de novo or inherited PAX2 mutation. Other risk factors include teratogen exposure during pregnancy and systemic diseases that compromise kidney or eye structures and functions.
-
PAX2 Mutation Frequency: PAX2 mutations are found in approximately 10% of children with unilateral or bilateral renal hypoplasia. They are also identified in 8% of individuals with nonsyndromic renal hypodysplasia and 4% of individuals with isolated familial focal segmental glomerulosclerosis.
-
PAX2 Mutation Hotspot: A mutational hotspot has been identified in the PAX2 gene, suggesting that specific regions of the gene are more prone to mutations. Germline mosaicism has also been reported, indicating that mutations can occur in parental germ cells.
Clinical Studies and Management
Clinical studies and effective management strategies are essential for improving the quality of life for individuals with PRS.
-
Clinical Studies: Several clinical studies have investigated the frequency of clinical symptoms and PAX2 mutations in pediatric patients. For example, a study in Japanese pediatric patients found that PAX2 mutations were associated with a range of renal and ocular abnormalities.
-
Management and Treatment: There is no specific treatment for PRS, and management is primarily supportive. Patients may require dialysis or kidney transplantation due to end-stage renal disease, while eye abnormalities may necessitate surgical intervention to prevent further complications.
Research and Future Directions
Ongoing research aims to better understand the molecular mechanisms underlying PRS and to identify other genes that may contribute to its development.
-
Research and Future Directions: Ongoing research aims to better understand the molecular mechanisms underlying PRS and to identify other genes that may contribute to its development. This knowledge could lead to more targeted therapeutic interventions and improved management strategies for affected individuals.
-
Variability in Manifestations: The manifestations of PRS can vary widely among individuals, suggesting that other unidentified genes may contribute to its development. This variety of manifestations is reflected in the naming of this collection of findings as a syndrome rather than a disease.
-
PAX2 Mutation Frequency: PAX2 mutations are found in approximately 10% of children with unilateral or bilateral renal hypoplasia. They are also identified in 8% of individuals with nonsyndromic renal hypodysplasia and 4% of individuals with isolated familial focal segmental glomerulosclerosis.
Final Thoughts on Papillorenal Syndrome
Papillorenal syndrome, also known as renal coloboma syndrome, is a rare genetic disorder that affects both the kidneys and eyes. Caused by mutations in the PAX2 gene, this condition can lead to serious complications like end-stage renal disease and significant visual impairment. While the exact prevalence remains unclear, the syndrome's impact on affected individuals is profound. Symptoms can vary widely, making diagnosis and management challenging. Genetic testing plays a crucial role in identifying the disorder, and supportive care is essential for managing symptoms. Research continues to uncover more about the molecular mechanisms behind PRS, aiming to improve treatment options. Understanding this complex condition is vital for providing better care and support to those affected. Further studies may lead to more effective interventions, offering hope for improved outcomes in the future.
Frequently Asked Questions
Was this page helpful?
Our commitment to delivering trustworthy and engaging content is at the heart of what we do. Each fact on our site is contributed by real users like you, bringing a wealth of diverse insights and information. To ensure the highest standards of accuracy and reliability, our dedicated editors meticulously review each submission. This process guarantees that the facts we share are not only fascinating but also credible. Trust in our commitment to quality and authenticity as you explore and learn with us.