


What is Pantothenate Kinase-Associated Neurodegeneration (PKAN)? PKAN is a rare, inherited brain disorder that leads to progressive movement problems, muscle stiffness, and cognitive decline. It affects about 1 to 3 people per million worldwide. Caused by mutations in the PANK2 gene, PKAN disrupts vitamin B5 metabolism, leading to iron buildup in the brain. Symptoms usually start between ages 7 and 15 but can appear at any age. There are two forms: classical, which progresses quickly, and atypical, which is slower. Diagnosis often involves genetic testing and MRI scans showing the "eye-of-the-tiger" sign. Currently, no cure exists, but treatments can help manage symptoms.
Key Takeaways:
- PKAN is a rare, inherited neurological disorder causing movement difficulties, dementia, and vision loss. It's diagnosed through genetic testing and MRI findings, with no cure but symptom management options available.
- PKAN is caused by mutations in the PANK2 gene, leading to brain iron accumulation. It can appear in childhood or adulthood, and community-specific mutations have been identified. Support from rare disease organizations and genetic testing are crucial for managing PKAN.
What is Pantothenate Kinase-Associated Neurodegeneration (PKAN)?
Pantothenate kinase-associated neurodegeneration (PKAN) is a rare, inherited neurological disorder. It involves the progressive degeneration of specific regions in the central nervous system. This condition falls under a broader group of disorders known as neurodegeneration with brain iron accumulation (NBIA).
-
Definition and Synonyms: PKAN is a genetic degenerative disease of the brain that can lead to parkinsonism, dystonia, dementia, and ultimately death. It is also known as neurodegeneration with brain iron accumulation type 1 (NBIA1) and formerly as Hallervorden-Spatz syndrome.
-
Incidence: The precise incidence of PKAN is unknown, but it is estimated to affect 1 to 3 per million people worldwide.
Genetic Basis and Inheritance
Understanding the genetic basis and inheritance pattern of PKAN can help in early diagnosis and management. This section delves into the genetic aspects of the disorder.
-
Genetic Basis: PKAN is caused by mutations in the PANK2 gene, which provides instructions for making the enzyme pantothenate kinase 2. These mutations lead to an inborn error of vitamin B5 (pantothenate) metabolism.
-
Inheritance Pattern: PKAN follows an autosomal recessive inheritance pattern. Both parents must be carriers of the mutated gene for their child to be affected. Each sibling of an affected individual has a 25% chance of being affected and a 50% chance of being an asymptomatic carrier.
Symptoms and Age of Onset
PKAN presents a variety of symptoms that can significantly impact the quality of life. The age at which these symptoms appear can vary widely.
-
Symptoms: The primary symptoms of PKAN include progressive difficulty with movement, involuntary muscle spasms, rigidity, and trouble with walking. Many people with this condition also develop problems with speech (dysarthria) and some may experience vision loss. Additionally, affected individuals may experience a loss of intellectual function (dementia) and psychiatric symptoms such as behavioral problems, personality changes, and depression.
-
Age of Onset: The disease typically presents between ages 7 and 15 years, but onset has been reported in all age groups, including infancy and adulthood.
Types of PKAN
PKAN can be classified into two main types: classical and atypical. Each type has its own set of characteristics and progression patterns.
- Classical vs. Atypical Forms: There are two subtypes of PKAN: classical and atypical. The classical form usually appears in early childhood, causing severe problems with movement that worsen rapidly. The atypical form appears later in childhood or adolescence and progresses more slowly. Signs and symptoms vary, but the atypical form is more likely than the classical form to involve speech defects and psychiatric problems.
Diagnosis and Pathophysiology
Diagnosing PKAN involves specific criteria and understanding its pathophysiology is crucial for developing effective treatments.
-
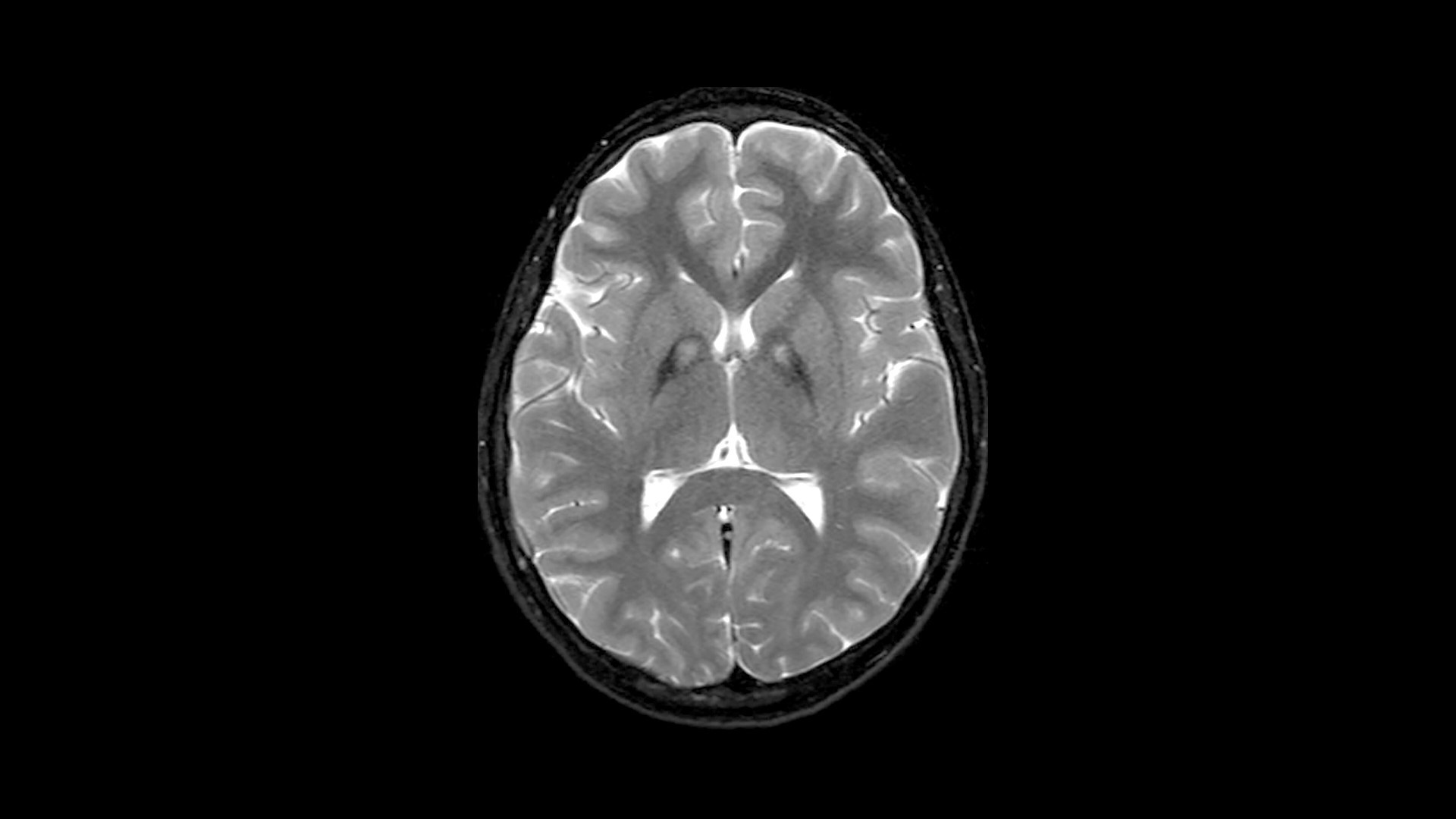
Diagnostic Criteria: PKAN is diagnosed by molecular genetic testing, most often after a characteristic finding on magnetic resonance imaging (MRI), called the “eye-of-the-tiger” sign, is detected. This sign indicates an accumulation of iron in the brain.
-
Pathophysiology: The disorder is characterized by an abnormal buildup of iron in specific areas of the brain. The PANK2 enzyme is crucial for converting vitamin B5 into coenzyme A, which is essential for energy and lipid metabolism. Disruption of this enzyme leads to accumulation of potentially harmful compounds in the brain, including iron.
Brain Iron Accumulation and MRI Findings
Iron accumulation in the brain is a hallmark of PKAN. MRI findings play a significant role in diagnosing the disorder.
-
Brain Iron Accumulation: The primary pathophysiological feature of PKAN is the accumulation of iron in the brain, particularly in the basal ganglia and globus pallidus. This iron accumulation is associated with neurodegeneration and the development of extrapyramidal symptoms such as parkinsonism and dystonia.
-
MRI Findings: Almost all cases of PKAN show an “eye-of-the-tiger” sign on MRI scans, which is indicative of iron accumulation in the brain. This sign is a characteristic feature used for diagnosis.
Genetic Mutations and Community-Specific Cases
Mutations in the PANK2 gene are the root cause of PKAN. Some communities have specific mutations that are more prevalent.
-
PANK2 Gene Location: The PANK2 gene is located on chromosome 20p13-p12.3. Mutations in this gene lead to the loss of function of the pantothenate kinase enzyme, which is essential for coenzyme A synthesis.
-
Mutations in PANK2 Gene: Mutations in the PANK2 gene can be null or missense mutations. A notable mutation is a 7bp deletion in the PANK2 gene coding sequence. These mutations disrupt the enzyme's function, leading to the accumulation of harmful compounds in the brain.
-
Community-Specific Mutations: PKAN has been reported in specific communities based on intra-community marriages where both parents carry the same mutation. One such community is the Agrawal (Agarwal) community mainly based in Northern India. The known mutation in this community is pathogenic mutation 1c.215_216insA.
Historical Background and Name Change
The history of PKAN includes its initial discovery and subsequent name change due to ethical concerns.
-
Historical Background: The disease was first described in 1922 by two German physicians, Hallervorden and Spatz, as a form of familial brain degeneration characterized by cerebral iron deposition.
-
Name Change: The name Hallervorden-Spatz syndrome was discouraged due to concerns regarding Dr. Hallervorden’s and Dr. Spatz’s affiliation with the Nazi regime and their unethical activities surrounding autopsy specimens. The condition is now more commonly referred to as pantothenate kinase-associated neurodegeneration (PKAN).
Treatment and Symptom Management
Currently, there is no cure for PKAN, but various treatments can help manage symptoms and improve quality of life.
-
Lack of Treatment: Currently, there are no treatments that modify the disease's progress. However, various medications and therapies can help improve symptoms. Active research is ongoing to develop new treatments.
-
Symptom Management: Management of PKAN focuses on symptom relief rather than halting disease progression. Medications such as anticholinergics, benzodiazepines, and dopaminergic agents may be used to manage symptoms like dystonia and parkinsonism.
Support and Genetic Testing
Support from rare disease organizations and genetic testing are crucial for managing PKAN.
-
Rare Disease Organizations: Organizations like the National Organization for Rare Disorders (NORD) provide assistance programs for patients with PKAN. These programs include the RareCare assistance program, MedicAlert assistance program, Rare Disease Educational Support Program, and Rare Caregiver Respite Program.
-
Genetic Testing: Genetic testing is crucial for diagnosing PKAN. It involves identifying mutations in the PANK2 gene. This testing can be performed through various methods including whole exome sequencing.
Clinical Features and Related Syndromes
PKAN has distinct clinical features and is related to other syndromes like HARP.
-
Clinical Features: Clinical features of PKAN include progressive extrapyramidal symptoms, dysarthria, and cognitive decline. The disease can also lead to psychiatric symptoms such as behavioral problems and depression.
-
HARP Syndrome: Historically, a condition called HARP (hypoprebetalipoproteinemia, acanthocytosis, retinitis pigmentosa, and pallidal degeneration) syndrome was described as a separate syndrome. However, it is now considered part of PKAN.
Research and Global Prevalence
Ongoing research aims to develop new treatments for PKAN. The global prevalence of the disorder remains low.
-
Research and Development: Ongoing research aims to understand the pathophysiology of PKAN better and to develop targeted therapies. Studies have shown that cysteine accumulation and iron chelation play significant roles in the disease's progression.
-
Global Prevalence: The accurate prevalence of PKAN is unclear, particularly in the Chinese population. However, it is estimated to be extremely low, affecting approximately 1–3 per million people globally.
Community Awareness
Raising awareness about PKAN within specific communities is crucial for early diagnosis and management.
- Community Awareness: Raising awareness about PKAN within specific communities where the disease is more prevalent is crucial for early diagnosis and management. This includes educating healthcare providers about the disease and its diagnostic criteria.
Final Thoughts on PKAN
Pantothenate kinase-associated neurodegeneration (PKAN) is a rare, inherited disorder marked by progressive brain degeneration and iron buildup. Affecting roughly 1 to 3 per million people, PKAN stems from mutations in the PANK2 gene. Symptoms range from movement difficulties and muscle spasms to speech issues and vision loss. Diagnosis often involves genetic testing and MRI scans showing the “eye-of-the-tiger” sign. While no cure exists, symptom management through medications and therapies can improve quality of life. Ongoing research aims to develop targeted treatments. Raising awareness, especially in communities with higher prevalence, is crucial for early diagnosis and better management. Organizations like NORD offer support programs for those affected. Understanding PKAN’s genetic basis and clinical features helps in providing appropriate care and advancing research efforts.
Frequently Asked Questions
Was this page helpful?
Our commitment to delivering trustworthy and engaging content is at the heart of what we do. Each fact on our site is contributed by real users like you, bringing a wealth of diverse insights and information. To ensure the highest standards of accuracy and reliability, our dedicated editors meticulously review each submission. This process guarantees that the facts we share are not only fascinating but also credible. Trust in our commitment to quality and authenticity as you explore and learn with us.