


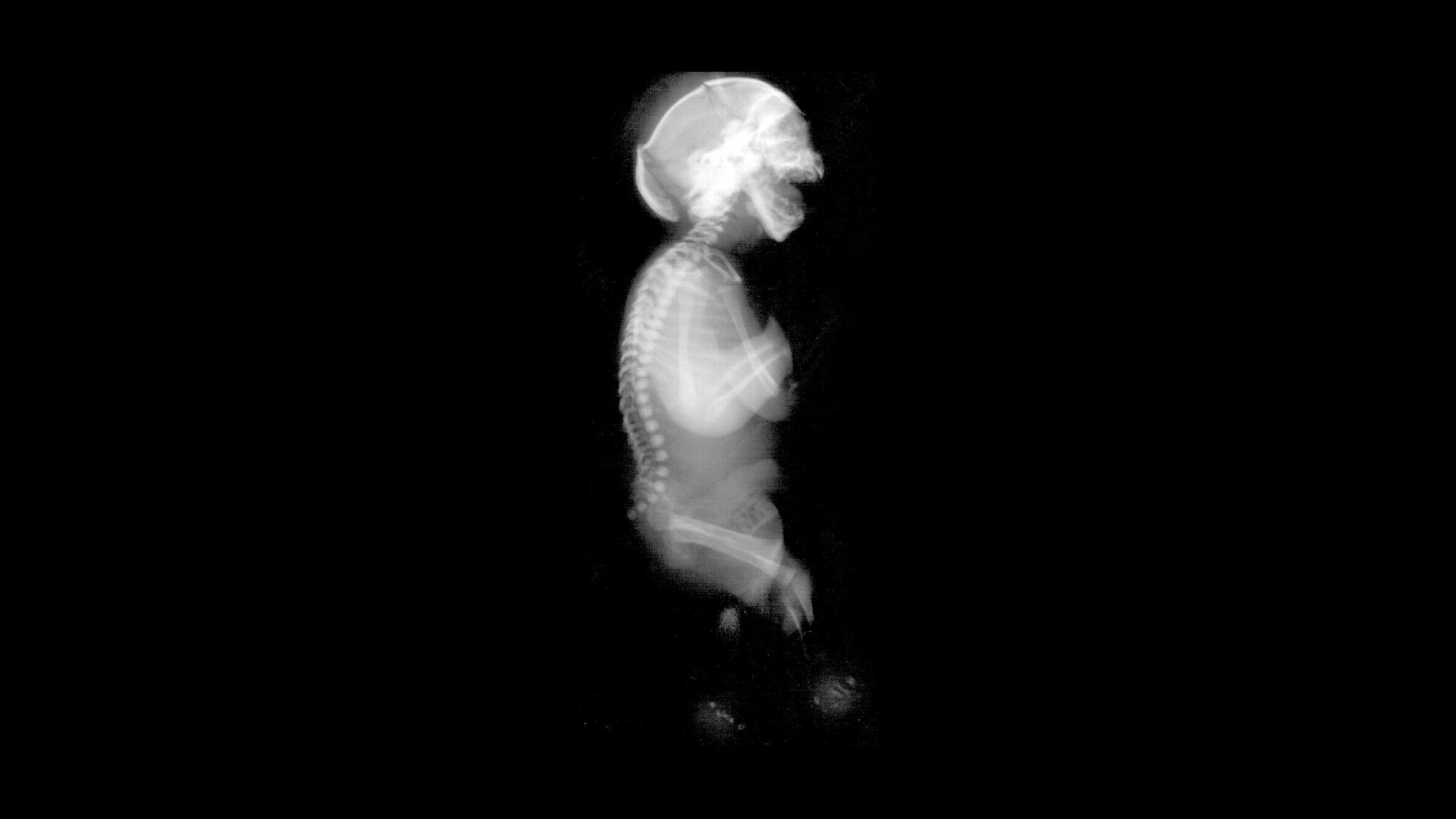
What is Neu-Laxova Syndrome? Neu-Laxova Syndrome (NLS) is a rare, severe genetic disorder marked by multiple congenital malformations and significant intrauterine growth restriction. First identified in the early 1970s by Dr. Richard Neu and Dr. Renata Laxova, this condition affects fewer than 100 reported cases worldwide. NLS is inherited in an autosomal recessive manner, requiring both parents to carry the mutated gene. The syndrome results from mutations in genes involved in the serine biosynthesis pathway, crucial for cell growth. Affected individuals often exhibit distinct facial features, skin abnormalities, limb malformations, and central nervous system anomalies. Tragically, most affected infants do not survive beyond the neonatal period.
Key Takeaways:
- Neu-Laxova Syndrome is an extremely rare and severe genetic disorder, affecting multiple body systems. It has no cure, and affected individuals often have a poor prognosis, with most being stillborn or dying shortly after birth.
- Understanding the genetic cause, clinical features, and challenges of diagnosing Neu-Laxova Syndrome is crucial for managing the condition. Ongoing research provides hope for future advancements in treatment and diagnosis.
What is Neu-Laxova Syndrome?
Neu-Laxova syndrome (NLS) is a rare and severe genetic disorder. It affects multiple systems in the body, leading to significant health challenges. Here are some key facts to understand this condition better.
-
Definition and Rarity: Neu-Laxova syndrome is extremely rare, with fewer than 100 cases reported worldwide. It involves severe intrauterine growth restriction and multiple congenital malformations.
-
Autosomal Recessive Inheritance: This syndrome is inherited in an autosomal recessive manner. Both parents must carry the mutated gene for their child to be affected.
-
Genetic Cause: Mutations in the PHGDH, PSAT1, or PSPH genes cause Neu-Laxova syndrome. These genes are crucial for the serine biosynthesis pathway, essential for cell growth.
Clinical Features of Neu-Laxova Syndrome
The clinical presentation of NLS is quite striking and severe. Here are some of the main features observed in affected individuals.
-
Characteristic Facial Features: Individuals often have proptosis (bulging eyes), eyelid malformations, a round and gaping mouth, micrognathia (small jaw), and low-set or malformed ears.
-
Facial Dysmorphism: The face appears severely distorted with features like proptosis, hypertelorism (wide-set eyes), and a flat nose.
-
Skin Abnormalities: Severe skin issues such as ichthyosis (dry, scaly skin) and hyperkeratosis (thickened outer skin layer) are common.
-
Limb Malformations: Limb issues include syndactyly (fused fingers or toes), contractures, and other deformities.
-
Central Nervous System (CNS) Malformations: CNS problems include microcephaly (small head), lissencephaly (smooth brain surface), microgyria (abnormal brain folding), cerebellum hypoplasia, and agenesis of the corpus callosum.
Prenatal and Postnatal Diagnosis
Diagnosing Neu-Laxova syndrome can be challenging but is crucial for managing the condition. Here’s how it is typically diagnosed.
-
Intrauterine Growth Restriction (IUGR): Severe IUGR is a hallmark of NLS, often leading to polyhydramnios (excessive amniotic fluid).
-
Generalized Edema: Generalized swelling and flexion deformities are often present, causing massive swelling of the scalp, joints, hands, and feet.
-
Prenatal Diagnosis: Ultrasound in the second trimester can reveal signs like polyhydramnios, IUGR, microcephaly, proptosis, and decreased fetal movement.
-
Genetic Study: Confirming the diagnosis involves genetic studies of amniotic fluid or blood samples, identifying mutations in PHGDH, PSAT1, or PSPH genes.
Treatment and Prognosis
There is no cure for Neu-Laxova syndrome, but understanding the prognosis and potential treatments can help manage the condition.
-
Treatment: No specific treatment exists, but serine and glycine supplementation has shown tentative benefits in related conditions.
-
Prognosis: The outlook is poor, with most affected individuals being stillborn or dying shortly after birth. The longest reported survival is 134 days.
-
Recurrence Risk: If both parents are carriers, there is a 25% chance of recurrence in future pregnancies.
Demographics and Research
Understanding who is affected and ongoing research can provide hope for future advancements.
-
Ethnic Background: NLS has been reported in various ethnic backgrounds, indicating it is not limited to any specific population.
-
Consanguinity: About 42% of cases involve parental consanguinity, suggesting a higher risk in families with consanguineous marriages.
-
Histopathological Findings: Histopathological features include ichthyosis, massive fat with hypertrophy of fat cells, and edema. Poor cortical bone formation and CNS anomalies are also observed.
-
Ultrasound Findings: Routine ultrasounds at 19-20 weeks may show polyhydramnios, IUGR, hypoechogenic skeletal structures, microcephaly, prominent eyes, retrognathism, hypomobility with flexion deformities, and massive swelling of the scalp, knee, elbow joints, hands, and feet.
Classification and Research Activities
Neu-Laxova syndrome is a complex condition that requires ongoing research to understand better and manage.
-
Clinical Genetics Review: NLS is classified under clinical genetics reviews due to its complex nature, characterized by neuro-oculo-ectodermal dysplasia, including IUGR, facial dysmorphism, and CNS malformations.
-
Research Activities: Research is ongoing to understand the causes and identify potential treatments. Gene mapping studies have pinpointed mutations in PHGDH, PSAT1, and PSPH as primary causes.
-
Newborn Screening: NLS is not part of standard newborn screening programs due to its rarity and lack of specific diagnostic markers. Advances in genetic testing may soon allow for prenatal or birth screening.
Patient Resources and Mild Forms
Support and information for affected families are crucial, even though resources are limited.
-
Patient-Centered Resources: Due to its rarity, resources are limited. Organizations like Orphanet provide detailed information on clinical features, diagnosis, and management options.
-
Mild Forms: Some cases of NLS are milder, with affected individuals surviving beyond infancy. These cases often have fewer severe malformations but still exhibit characteristic features like ichthyosis and limb anomalies.
Historical Context
Understanding the history of Neu-Laxova syndrome helps appreciate the progress made in recognizing and diagnosing this condition.
- Historical Context: First described in the early 1970s by Dr. Richard Neu and Dr. Renata Laxova, approximately 72 cases have been reported in medical literature, highlighting the rarity and severity of this condition.
Final Thoughts on Neu-Laxova Syndrome
Neu-Laxova Syndrome is a rare, severe genetic disorder marked by multiple congenital malformations and significant intrauterine growth restriction. With fewer than 100 cases reported, it's caused by mutations in the PHGDH, PSAT1, or PSPH genes. This condition leads to distinct facial features, skin abnormalities, limb malformations, and central nervous system issues. Prenatal diagnosis is possible through ultrasound and genetic testing. Unfortunately, there's no specific treatment, and the prognosis remains poor, with most affected individuals not surviving beyond infancy. Understanding the genetic causes and clinical features is crucial for providing accurate information to families and healthcare providers. While research continues, the rarity of Neu-Laxova Syndrome makes it a challenging condition to study and treat. Awareness and ongoing research are key to improving our knowledge and potentially finding therapeutic interventions in the future.
Frequently Asked Questions
Was this page helpful?
Our commitment to delivering trustworthy and engaging content is at the heart of what we do. Each fact on our site is contributed by real users like you, bringing a wealth of diverse insights and information. To ensure the highest standards of accuracy and reliability, our dedicated editors meticulously review each submission. This process guarantees that the facts we share are not only fascinating but also credible. Trust in our commitment to quality and authenticity as you explore and learn with us.