

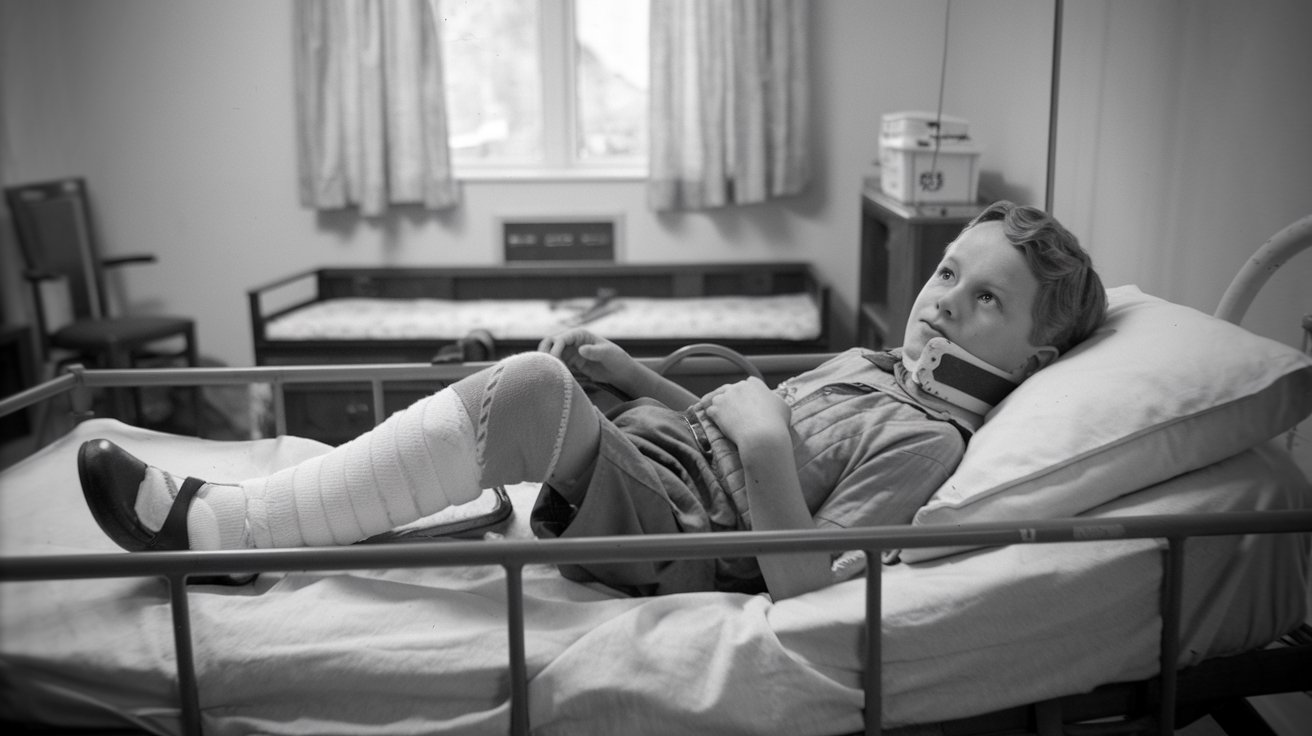
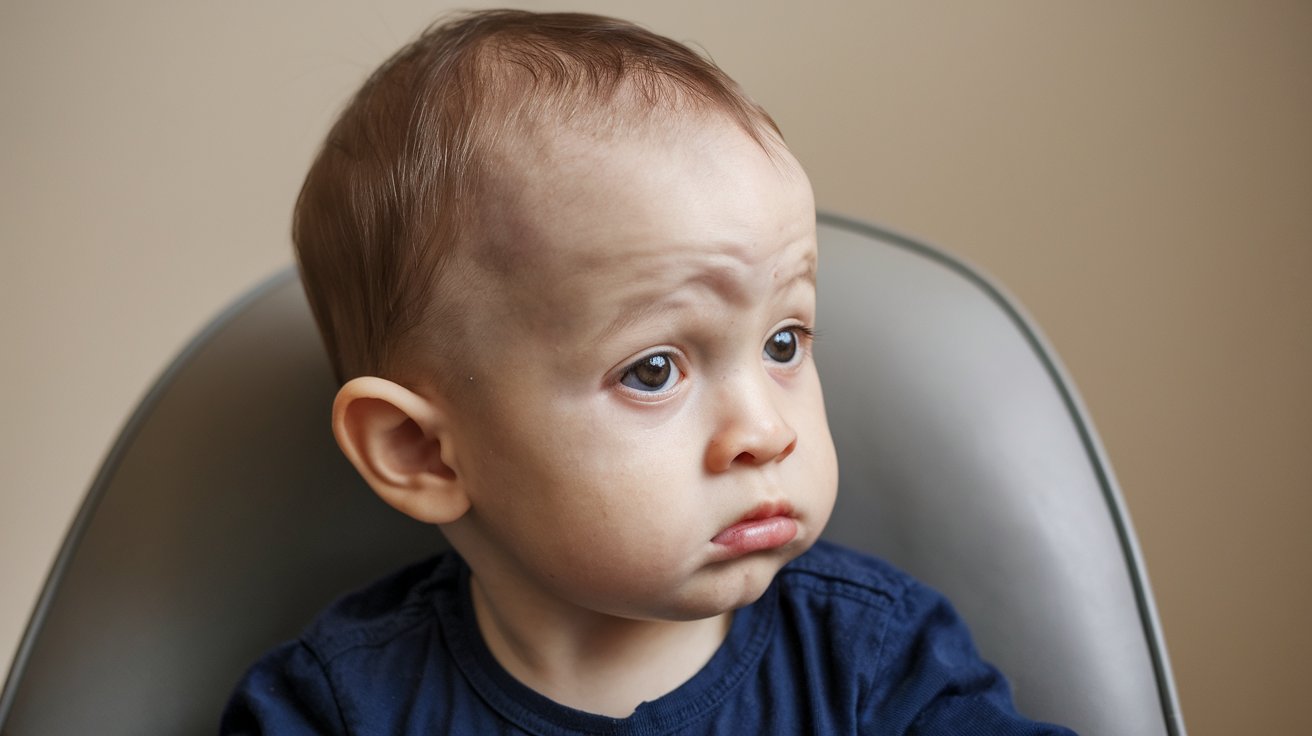
What is Langer-Giedion Syndrome? Langer-Giedion Syndrome (LGS) is a rare genetic disorder caused by a deletion on chromosome 8. This condition, also known as trichorhinophalangeal syndrome type II (TRPS2), affects various parts of the body, leading to unique facial features, skeletal abnormalities, and developmental issues. People with LGS often have short stature, mild to moderate learning difficulties, and distinctive facial characteristics like a long philtrum and bulbous nasal tip. The syndrome is autosomal dominant, meaning only one copy of the mutated gene is needed to cause the condition. Despite its rarity, understanding LGS is crucial for early diagnosis and effective management.
What is Langer-Giedion Syndrome?
Langer-Giedion Syndrome (LGS) is a rare genetic disorder. It involves a deletion on chromosome 8, leading to various developmental issues. This condition is also known as trichorhinophalangeal syndrome type II (TRPS2).
-
Definition and Synonyms: LGS is caused by a deletion on chromosome 8, specifically in the region 8q24.1. It's also called TRPS2 and the Langer-Giedion chromosome region (LGCR).
-
Causes: The syndrome results from a missing piece of chromosome 8, which includes the TRPS1 gene. This gene is crucial for normal development.
-
Inheritance Pattern: LGS is an autosomal dominant disorder. Only one copy of the mutated gene is needed to cause the condition. Affected individuals have a 50% chance of passing it to their children.
Symptoms of Langer-Giedion Syndrome
LGS presents a variety of symptoms, ranging from mild to severe. These symptoms can affect physical appearance, growth, and development.
-
Mild to Moderate Learning Difficulties: Many individuals with LGS experience learning difficulties that can range from mild to moderate.
-
Short Stature: Progressive growth retardation often leads to short stature in those with LGS.
-
Unique Facial Features: Characteristic facial features include a long prominent philtrum, thin upper lip, wide-spaced eyes, bulbous nasal tip, broad nasal bridge, wide nostrils, micrognathia, retrognathia, deep-set eyes, and large ears.
-
Skeletal Abnormalities: Skeletal issues include cone-shaped epiphyses of the hand phalanges and short fingers and toes. The fifth fingers may be bent. Other skeletal problems include winged scapula, thin ribs, and scoliosis.
-
Exostoses: Benign bony growths called exostoses often develop, which can cause complications like spinal cord compression, asymmetric limb growth, and reduced mobility.
-
Joint Hypermobility: Initially, joint hypermobility is common, but it progresses to joint stiffness later in life when osteochondromas develop.
-
Hip Dysplasia: Hip dysplasia may develop, usually in early adulthood, though it can occur in infancy or childhood.
-
Skin, Hair, Sweat Glands, and Nails: Ectodermal dysplasia is a key feature. Most individuals have sparse scalp hair, particularly severe in males who often experience alopecia shortly after puberty. However, eyebrows may be unusually thick.
Additional Complications and Diagnosis
LGS can also lead to other complications and requires specific diagnostic methods for confirmation.
-
Dental Abnormalities: Dental issues such as extra central incisors and missing teeth may occur.
-
Developmental Issues: Affected individuals may exhibit mild to severe mental retardation, hearing loss (sensorineural deafness), and delayed speech development. The range and severity of symptoms vary greatly.
-
Diagnosis: Diagnosis is usually made at birth or early childhood. Genetic testing confirms the deletion on chromosome 8. Imaging studies like X-rays and MRIs assess skeletal abnormalities and exostoses.
-
Differential Diagnosis: LGS can be differentiated from other conditions like tricho-rhino-phalangeal syndrome type 1, Fibrodysplasia Ossificans Progressiva, trichorhinophalangeal syndrome type 3, multiple exostoses, and Legg-Calvé-Perthes disease.
Managing Langer-Giedion Syndrome
Managing LGS involves a multidisciplinary approach to address the various symptoms and complications.
-
Genetic Testing: Genetic testing identifies the deletion on chromosome 8 and confirms the absence of the TRPS1 gene. Methods like PCR (Polymerase Chain Reaction) and FISH (Fluorescence In Situ Hybridization) are used.
-
Management: Management includes orthopedic care for skeletal abnormalities and exostoses, speech therapy for delayed speech development and hearing loss, educational support for learning difficulties, physical therapy for joint mobility, and psychological support for emotional and psychological impact.
-
Prognosis: Prognosis varies depending on symptom severity. Regular medical follow-up and early intervention can significantly improve the quality of life.
-
Prevalence: LGS is extremely rare, estimated to occur in fewer than 1 in 100,000 births.
-
Research and Future Directions: Ongoing research aims to better understand the genetic and molecular mechanisms of LGS. Future directions may involve developing more accurate diagnostic tools and improving management strategies.
Final Thoughts on Langer-Giedion Syndrome
Langer-Giedion syndrome, also known as trichorhinophalangeal syndrome type II (TRPS2), is a rare genetic disorder caused by a deletion on chromosome 8. This condition presents a variety of symptoms, including unique facial features, skeletal abnormalities, and learning difficulties. Diagnosis often involves genetic testing to confirm the deletion. Managing LGS requires a multidisciplinary approach, including orthopedic care, speech therapy, and educational support. While the syndrome is rare, affecting fewer than 1 in 100,000 births, understanding its complexities can significantly improve the quality of life for those affected. Continued research is essential for developing better diagnostic tools and management strategies. By staying informed and proactive, families and healthcare providers can offer the best possible care for individuals with Langer-Giedion syndrome.
Was this page helpful?
Our commitment to delivering trustworthy and engaging content is at the heart of what we do. Each fact on our site is contributed by real users like you, bringing a wealth of diverse insights and information. To ensure the highest standards of accuracy and reliability, our dedicated editors meticulously review each submission. This process guarantees that the facts we share are not only fascinating but also credible. Trust in our commitment to quality and authenticity as you explore and learn with us.